Role of Ubiquitinationin Hepcidin-Mediated FerroportinInternalizationIntroduction
Abstract
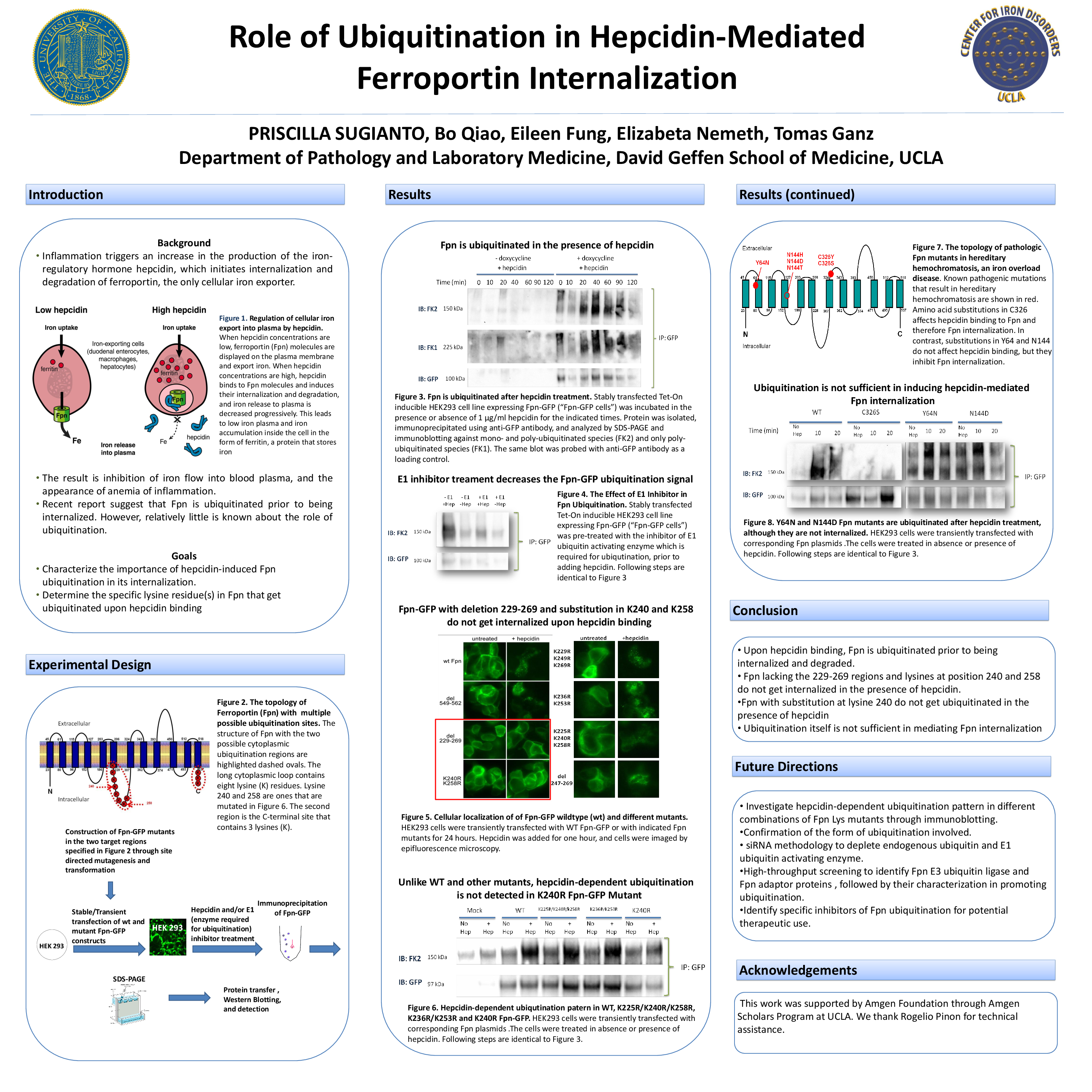
Anemia of inflammation is one of the most common immunopathological disorders that affect patients with acute and chronic infections or inflammatory diseases. Inflammation causes increased production of hepcidin, which binds to the only cellular iron exporter ferroportin and induces its internalization and degradation, thus blocking iron efflux from cells into plasma. This then restricts iron flow to the developing red cells in bone marrow, limits hemoglobin synthesis and results in anemia of inflammation. Preliminary studies have shown that ferroportin is ubiquitinated prior to its internalization, but it is still unknown where ferroportin is ubiquitinated and whether this modification is necessary or sufficient for ferroportin internalization. The goal of this study was to characterize the sites of hepcidin-induced ferroportin ubiquitination and the effect of ubiquitination on ferroportin internalization. A number of ferroportin mutants were constructed by site directed mutagenesis and analyzed for hepcidin-induced (by fluorescence microscopy) and for ubiquitination (by immunoblotting). Upon hepcidin treatment, ferroportin with substitution in lysine 240 and 258 was not internalized and one with substitution in lysine 240 was not ubiquitinated. Lastly, we detected that pathogenic ferroportin mutants which were resistant to hepcidin and were not internalized, did get ubiquitinated. Our result suggests that lysines 240 and 258 are the two important sites for ferroportin ubiquitination and that ubiquitination is necessary, but not sufficient for its hepcidin-mediated internalization. Better understanding of the structural basis and the role of the hepcidin-mediated ferroportin ubiqutination and internalization should facilitate the rational design for the treatments of anemia of inflammation.
Related articles